The role of the TET2 DNA demethylase in cardiomyocyte hypertrophy and cardiomyopathy
Cardiovascular Research


Abstract
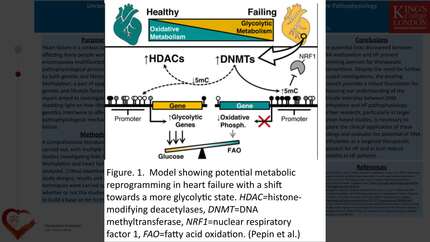
Heart failure (HF) represents the terminal outcome of pathological cardiac remodeling, a condition distinct from physiological adaptation1. Underlying these phenotypic alterations is an aberrant reprogramming of gene expression governed by epigenetic mechanisms. While methylation typically represses transcription, Ten-Eleven Translocation (TET) enzymes catalyze the oxidation of 5-methylcytosine (5mC) to 5-hydroxymethylcytosine (5hmC)2. This active demethylation process facilitates the recruitment of transcriptional machinery and subsequent gene activation. Although alterations in DNA methylation are documented in end-stage human HF3, it remains unclear whether these epigenetic shifts—and the specific enzymes driving them—play a causal role in disease progression.
This project aims to uncover the specific role of TET2-mediated DNA demethylation in driving the development of cardiomyocyte hypertrophy.
The study investigated TET isoform expression in human heart tissues, comparing Young (20-44y) versus Old (65-99y) donors, and patients with Ischemic (ICM) or Dilated Cardiomyopathy (DCM) against age-matched non-failing controls. Mass spectrometry was performed to measure the hmC/C ratio. Neonatal rat ventricular myocytes (NRVMs) were treated with Endothelin-1 (ET-1) to establish an in vitro model of hypertrophy. Hypertrophy was evaluated via cell surface area measurement and protein synthesis rates. Gene expression was analyzed via RT-qPCR, and 5-hydroxymethylcytosine (5hmC) levels were measured using dot blot assay. siRNA-mediated knockdown of Tet2 was performed in NRVMs to determine causality.
Observations in human heart failure: TET2 expression was significantly upregulated in ICM and DCM tissues compared to age-matched controls. This upregulation positively correlated with hypertrophic markers, such as NPPA and the MYH7/MYH6 ratio. Mass spectrometry and gene expression analysis indicated an increased hmC/C ratio and a reactivation of the fetal gene program in hypertrophic myocardium. Mechanistic insights from in vitro model: ET-1 stimulation in NRVMs led to significant hypertrophy, increased global protein synthesis, and the reactivation of hypertrophic genes (Nppa, Acta1). This hypertrophic response was accompanied by a notable upregulation of Tet2 (at both mRNA and protein levels). ET-1 treatment significantly increased global 5hmC levels, indicating enhanced DNA demethylase activity. Crucially, siRNA knockdown of Tet2 attenuated the ET-1-induced upregulation of hypertrophic markers, confirming that Tet2 is required for the full hypertrophic response.
TET2 contributes to cardiomyocyte hypertrophy by promoting active DNA demethylation. The pathological stress leads to Tet2 upregulation, increased 5hmC generation, and the subsequent activation of a maladaptive fetal gene program, highlighting the TET2-5hmC axis as a potential therapeutic target for cardiac remodeling.
Contributors


You may be interested in